
Solving the Polymorph Problem
The process of drug development is often described via metaphor. It’s a marathon, not a sprint. A great big funnel. Or even a 1,000 piece jigsaw puzzle. Perhaps the best analog to drug development is a game of incomplete information like poker. Throughout the life of the drug program, high-stakes decisions must be made using limited experimental evidence. At no point is this more true than when it comes to crystal polymorphism and solid form selection.
Crystal polymorphism is the tendency of a substance (here, a drug molecule) to exist in distinct crystalline forms. Various polymorphs of the same molecule may have very different physical properties. Take acetaminophen (or paracetamol) for example. The commercial acetaminophen in your medicine cabinet contains the form I polymorph, in which the molecules arrange in a herringbone-like pattern. Alternatively, acetaminophen can also crystallize into the more planar form II polymorph (among others).
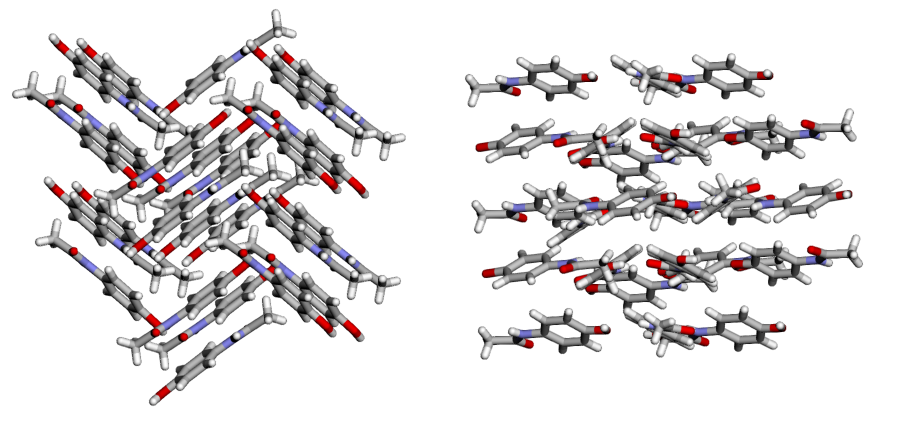 Left: Form I of acetaminophen. Right: Form II of acetaminophen
Left: Form I of acetaminophen. Right: Form II of acetaminophen
Manufacturers would rather produce form II, since the slip planes offer better compressibility, which makes for more durable tablets. Unfortunately for them, form II acetaminophen is less stable, and with time and temperature will convert to the more stable form I structure.
At this point, polymorphism might sound like a scientific curiosity, but it’s serious business. The canonical story of polymorphism-gone-wrong is the anti-viral drug ritonavir, first developed by Abbott in the late nineties as an HIV medicine under brand-name Norvir. Soon after Norvir hit the shelves, something was off. Scientists found that the ritonavir crystals, which were formulated as a particular polymorph (form I), were converting to a then-unknown and highly insoluble polymorph (now referred to as form II). This unexpected form II polymorph triggered a massive recall, leading to estimated loss of $250 million.
Understanding the polymorphic behavior of a drug molecule is therefore critical. However, it's not an easy process. Some drugs (like galunisertib) may have nearly a dozen known polymorphs, while others deterministically crystallize into a singular form. Because of this, drug developers must minimize the ever-present risk of a yet undiscovered polymorph. Historically, the best way to do this has been brute force laboratory experiments. A sample of the drug molecule is systematically crystallized under different experimental conditions—such as cooling from melt, solvent evaporation, anti-solvent addition, slurry equilibration, or mechanical grinding—across a range of temperatures, pressures, and solvents, as extensively as time and budgets permit.
Over the past few decades, computation has made some inroads into understanding polymorphism. The advent of this change was the adoption of Density Functional Theory (DFT), a modeling method that allows one to calculate properties of chemical systems from first principles. With DFT, one can compute properties of polymorphs (like relative stabilities) without ever having actually crystallized them in a lab. Paring DFT ranking with a method for generating plausible polymorphs allows for full-fledged polymorph prediction, commonly known as crystal structure prediction (CSP). Today, most pharmaceutical companies complement experimental polymorph screens with some kind of CSP.
At Lavo, we think there are huge gains to be made in modelling polymorphism, and we're developing new tools for drug developers. We're motivated by two beliefs. The first is that it's possible to do accurate CSP with far fewer DFT calculations (read: way less compute) than traditional approaches. This is made possible due to advances in machine learning for chemistry, a field in which Derek and I collaborated on throughout our PhDs. Below is a CSP result performed on the drug candidate LY-2624803, generated without any DFT calculations (only a pre-trained neural network is used). The calculation required roughly 6,000 CPU hours, which is drastically less than many other approaches.
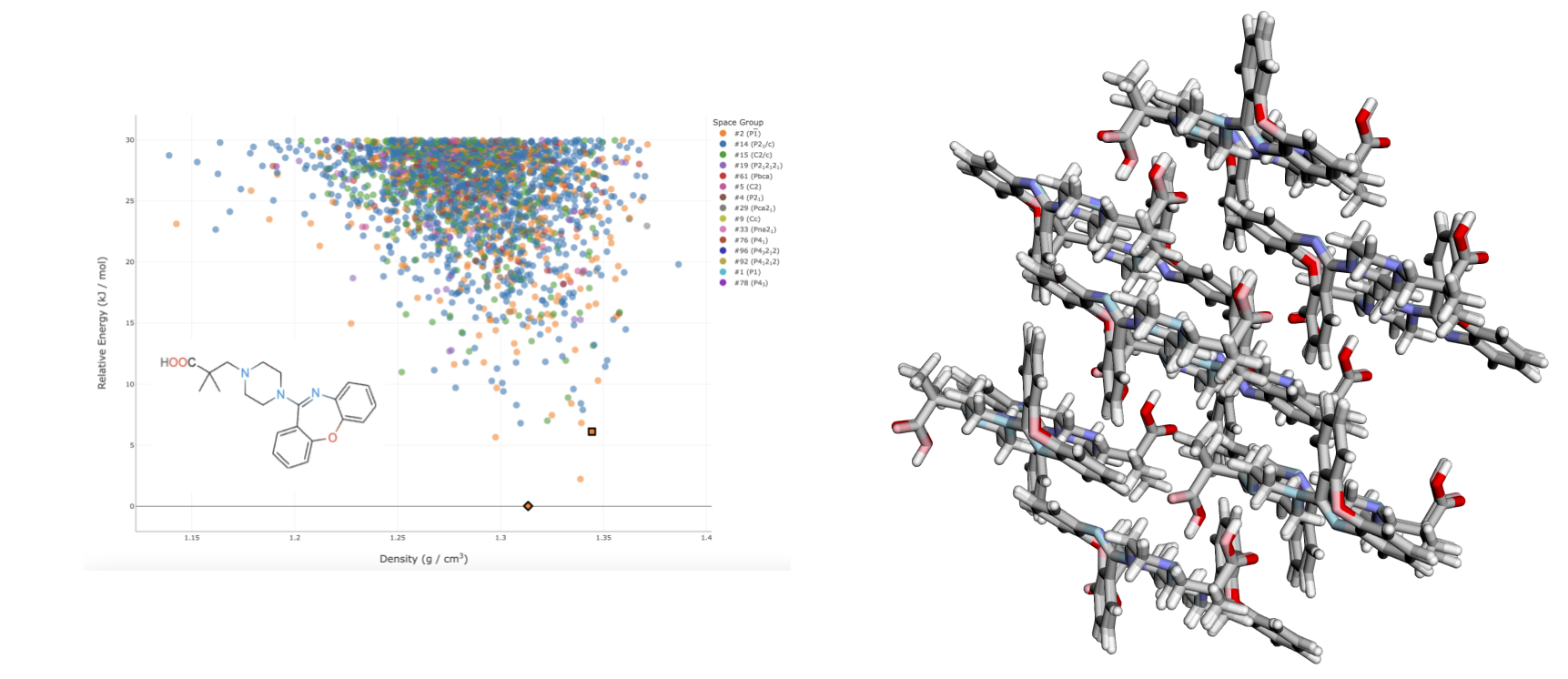 A CSP of LY-2624803. Left: the landscape of generated crystal structures, produced solely with a neural network potential. Matches to the two known experimental polymorphs are outlined. Right: an overlay of one of the generated crystal structures, and the matching experimentally determined crystal structure.
A CSP of LY-2624803. Left: the landscape of generated crystal structures, produced solely with a neural network potential. Matches to the two known experimental polymorphs are outlined. Right: an overlay of one of the generated crystal structures, and the matching experimentally determined crystal structure.
Lavo's second belief is that CSP should be an accessible, understandable tool for scientists to use. Traditional CSP requires deep technical knowledge of crystallography, chemistry, and high performance computing. It also often requires hands-on fine-tuning and human intuition. Wanting to overcome these limitations led us to develop the Crystal Console, a fully-managed, cloud-based web-platform designed specifically with ease-of-use in mind. Scientists no longer need their own specialized computing infrastructure; the Console handles all of the technical complexities behind the scenes, scaling effortlessly from exploratory screens to comprehensive polymorph predictions. Check out a video here, where the above CSP is performed in an afternoon with only a few clicks.
Interested in trying out the Console on your own drug molecules? Get in touch by emailing founders@lavo.ai.